
Cloud-native Bioinformatics: HPC to GCP
Working with a team from the Imperial College in London, we started by reviewing a bioinformatic analysis which is currently running on…

Working with a team from the Imperial College in London, we started by reviewing a genomic analysis workflow which is currently running on their HPC cluster. The workflow is a complete analysis for single-cell/nuclei RNA-sequencing data. I started by reviewing the workflow process steps generated by the Nextflow script used to run it.
Next I reviewed the current method of running this workflow by connecting with the team and observing them run this analysis on their HPC cluster. Of note is that the workflow utilizes containerization via Singularity / Docker.
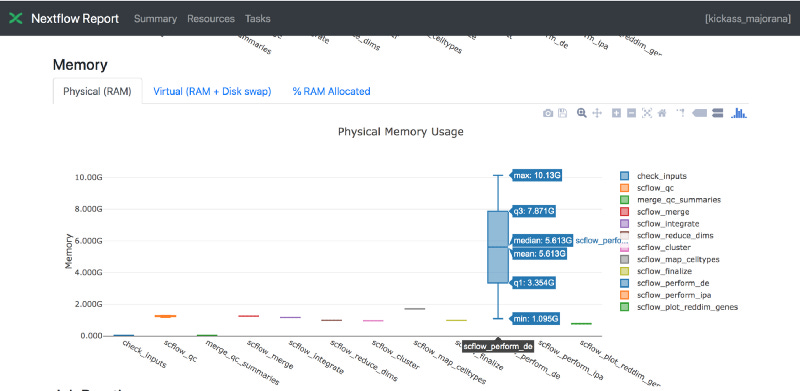
Also I asked the team to share run logs via visualizations using Nextflow Tower. I did so to quickly see which processes in the workflow had the highest resource needs. Shown below is one of the useful visualizations — showing Memory usage.
To The Cloud
A useful starting point when running any genomics workflow on the public cloud is to begin with a subsampled synthetic dataset. Real-world on premise workflows can run for hours, days or even weeks. To optimize for cloud, we will be more productive with fast feedback — i.e. did the workflow run correctly? how long did it take?…
15 minutes or less is the goal for each test.
To meet this goal we needed sample data. The Imperial College team created a ‘tiny’ (in relation to real-world work) example dataset by subsampling the open source mouse brain dataset.
Next we worked together so that I could understand the minimum machine type needed to run this workflow. In this case a key requirement is for 64 GB of RAM. We are working on GCP for this project, so I chose a GCE VM instance size of 32 CPUs and 120 GB of RAM. I started with a general purpose instance, but, due to the type of workload, will consider memory-optimized instances as work progresses. The instance I selected costs ~ 50 cents/hr. Of course using the ‘turn-if-off-when-not-in-use’ pattern for this VM helps to keep dev/test costs under control as well.
To get a first run, I simply modeled this powerful cloud-based VM as if it were a local machine. I created an appropriately-size GCE instance, based on a custom GCE/Nextflow test image that I had previously created which contained the required requisite software (JDK, Docker, Nextflow…) and updated the local.config file paths and started the analysis.
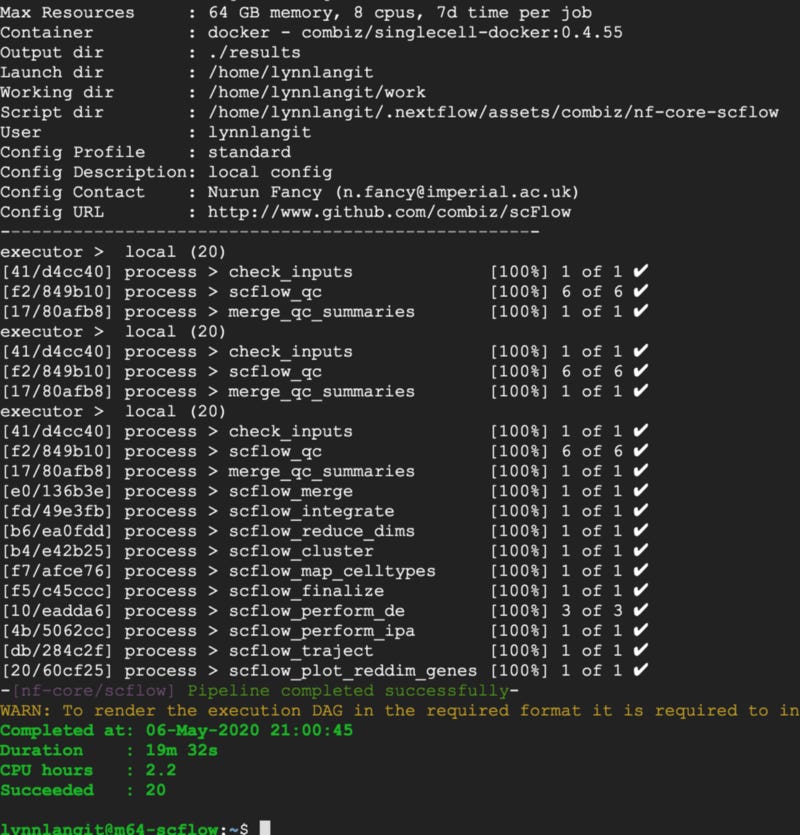
In 19 minutes…SUCCESS — output shown below. Note also the CPU hours for this run are shown at 2.2 hours.
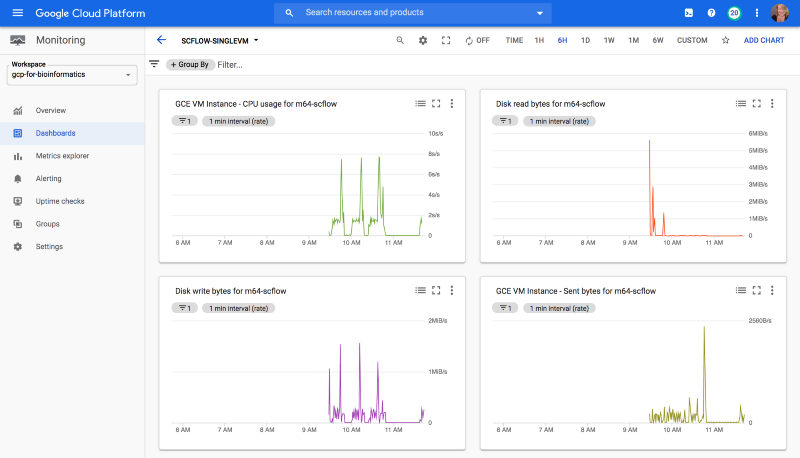
I created a quick dashboard for my test GCE VM using GCP Cloud Monitoring, so that I could get a sense of resource usage for this workflow.
From One to Many
Given that the example workflow used a tiny example dataset, it would be impractical to scale up to a single huge VM for real-world sized workloads. I doubt a such a massive singe instance even exists.
So the next step in moving scFlow to GCP is to use a managed cluster of VMs. There are a number of choices in how best to implement clustering for large-scale technical compute jobs on GCP. These include a number of choices in cluster-management software. These function similar to an on premise HPC cluster for high-throughput computing, i.e. using head nodes and compute nodes in a distributed fashion.
However, GCP includes an additional method of scaling out VM clusters — their Life Sciences API. From the GCP documentation:
The Cloud Life Sciences API is aimed at developers who want to build on or create job management tools (such as dsub) or workflow engines (such as Nextflow or Cromwell). The Cloud Life Sciences API provides a backend for these tools and systems, providing job scheduling for Docker-based tasks that perform secondary genomic analysis on Compute Engine containers.
The most common use case when using the Cloud Life Sciences API is to run an existing tool or custom script that reads and writes files, typically to and from Cloud Storage. The Cloud Life Sciences API can run independently over hundreds or thousands of these files.
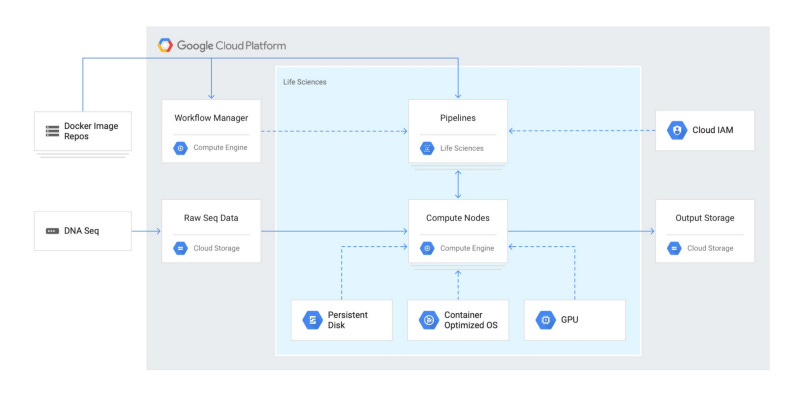
In order to evaluate this API for use in this scenario, I wanted to work through an example using Nextflow.
Life Sciences API and Nextflow
In Google’s example tutorial, an rna-seq pipeline is configured to run via the nextflow.config file with LS API. The example is a bit simpler than the scFlow pipeline that I am working with, but it’s a good starting point. Using the default settings, this took 10 minutes to run using cloud shell. Output is shown below.
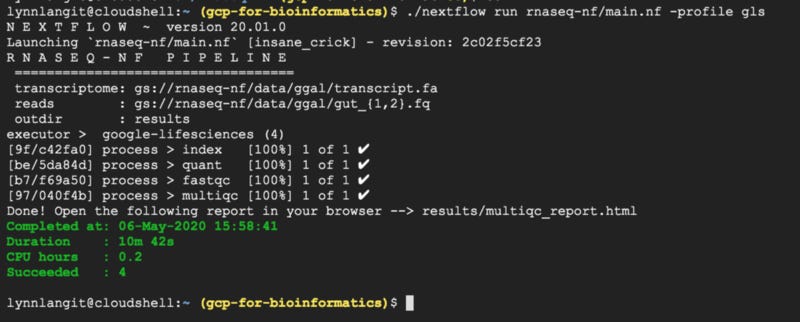
Of course the most interesting aspect of working with LS API, was the higher-level cluster management via the config file. In contrast to working with my alway-on-until-I-manually-turned-it-off test VM, using LS API, GCE VM instances were DYNAMICALLY allocated as needed by the processes in the workflow. These VMs were allocated, activated and de-activated automatically. They ran for a total of 0.2 CPU hours. Also these were small-sized VMs, 1 CPU and 1 GB RAM.
Of course these two analysis are NOT identical in size or complexity — however, scaling a 10 min run with 0.2 CPU hours vs. a 19 min run with 2.2 CPUs hours does strike me as quite significant.
Next Steps
Next I’ll be writing a nextflow.config file using LS API for the scFlow workflow and reporting back on CPU hours for the single VM vs. LS API run.